Rubinstein-Taybi Syndrome - GeneReviews® - NCBI Bookshelf
Por um escritor misterioso
Last updated 11 novembro 2024

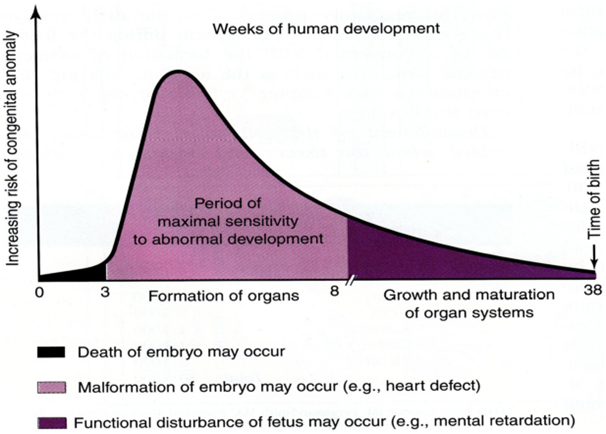
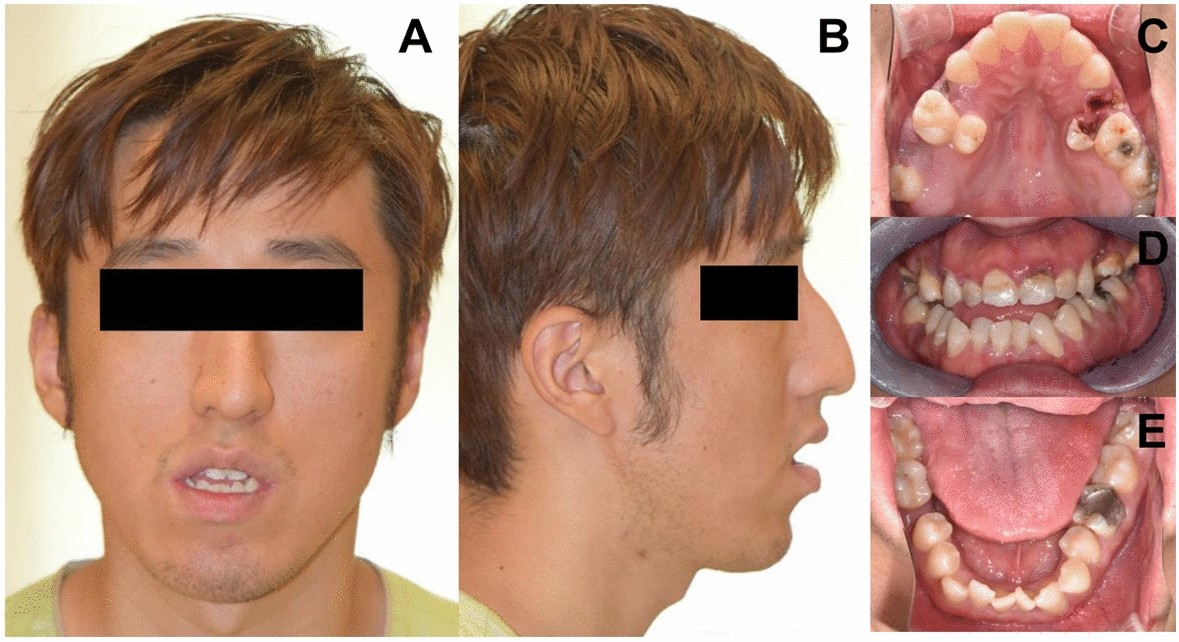
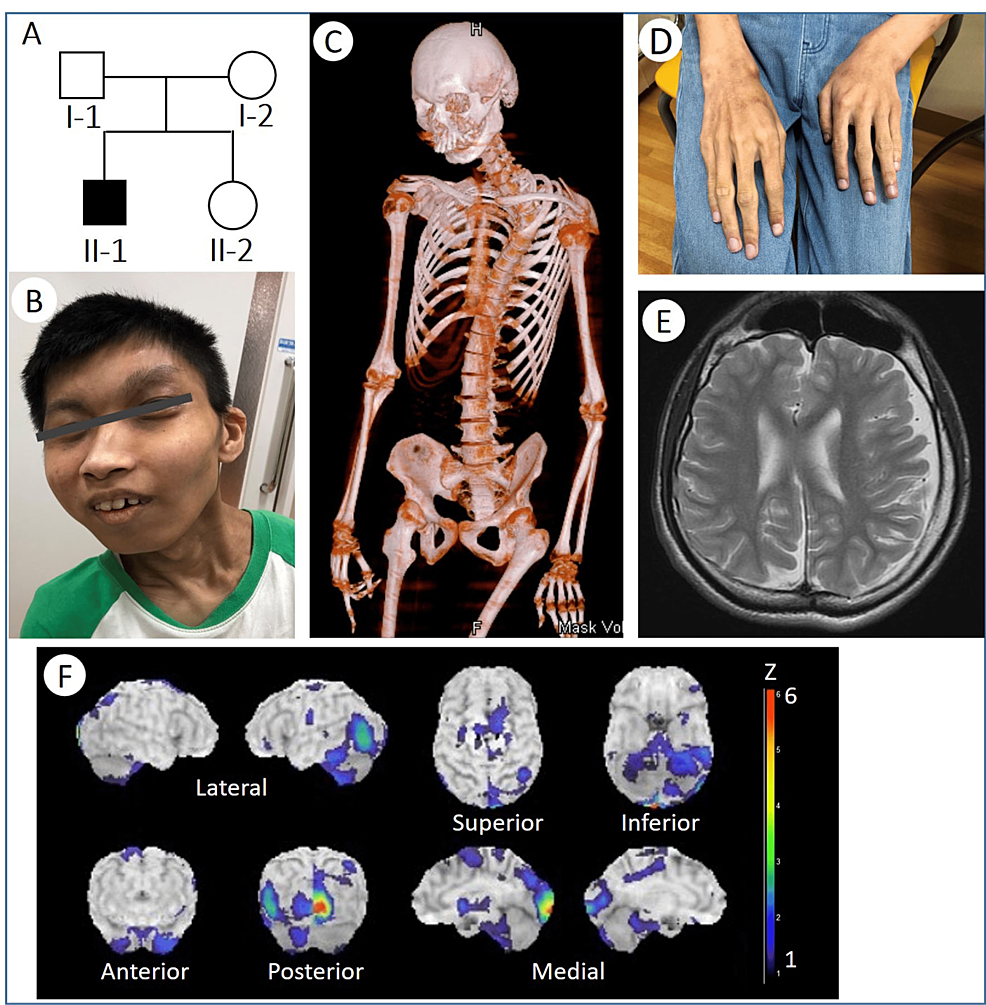
Rubinstein-Taybi syndrome (RSTS) is characterized by distinctive facial features, broad and often angulated thumbs and halluces, short stature, and moderate-to-severe intellectual disability. Characteristic craniofacial features include downslanted palpebral fissures, low-hanging columella, high palate, grimacing smile, and talon cusps. Prenatal growth is often normal, then height, weight, and head circumference percentiles rapidly drop in the first few months of life. Short stature is typical in adulthood. Obesity may develop in childhood or adolescence. Average IQ ranges between 35 and 50; however, developmental outcome varies considerably. Some individuals with EP300-related RSTS have normal intellect. Additional features include ocular abnormalities, hearing loss, respiratory difficulties, congenital heart defects, renal abnormalities, cryptorchidism, feeding problems, recurrent infections, and severe constipation.

Rubinstein-Taybi syndrome with scoliosis treated with single-stage posterior spinal fusion: illustrative case in: Journal of Neurosurgery: Case Lessons Volume 1 Issue 11 (2021) Journals
Pediatrics Neupsy Key

Chromosome 16p13.3 Deletion Syndrome, Proximal disease: Malacards - Research Articles, Drugs, Genes, Clinical Trials
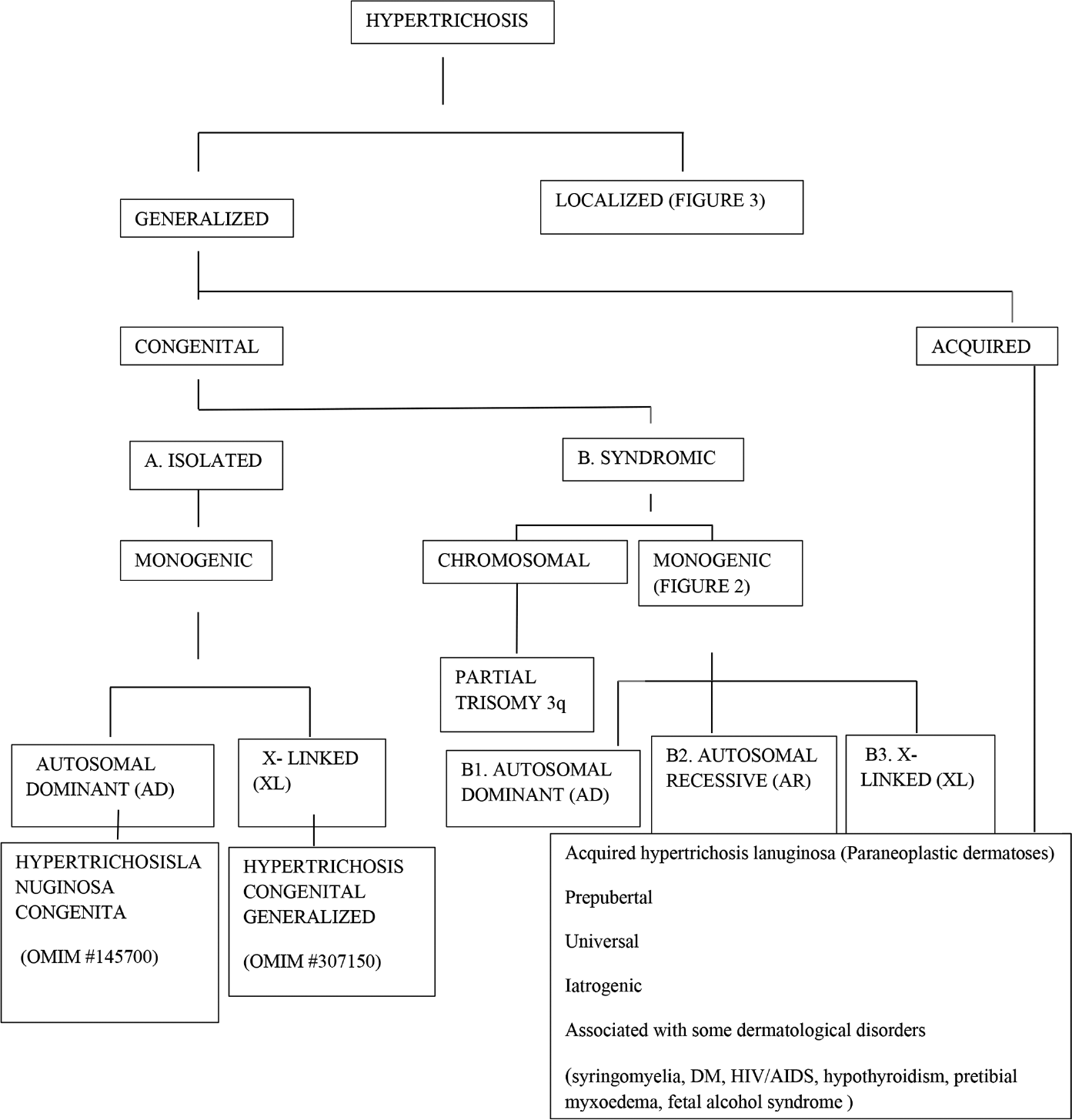
Approach to inherited hypertrichosis: A brief review - Indian Journal of Dermatology, Venereology and Leprology
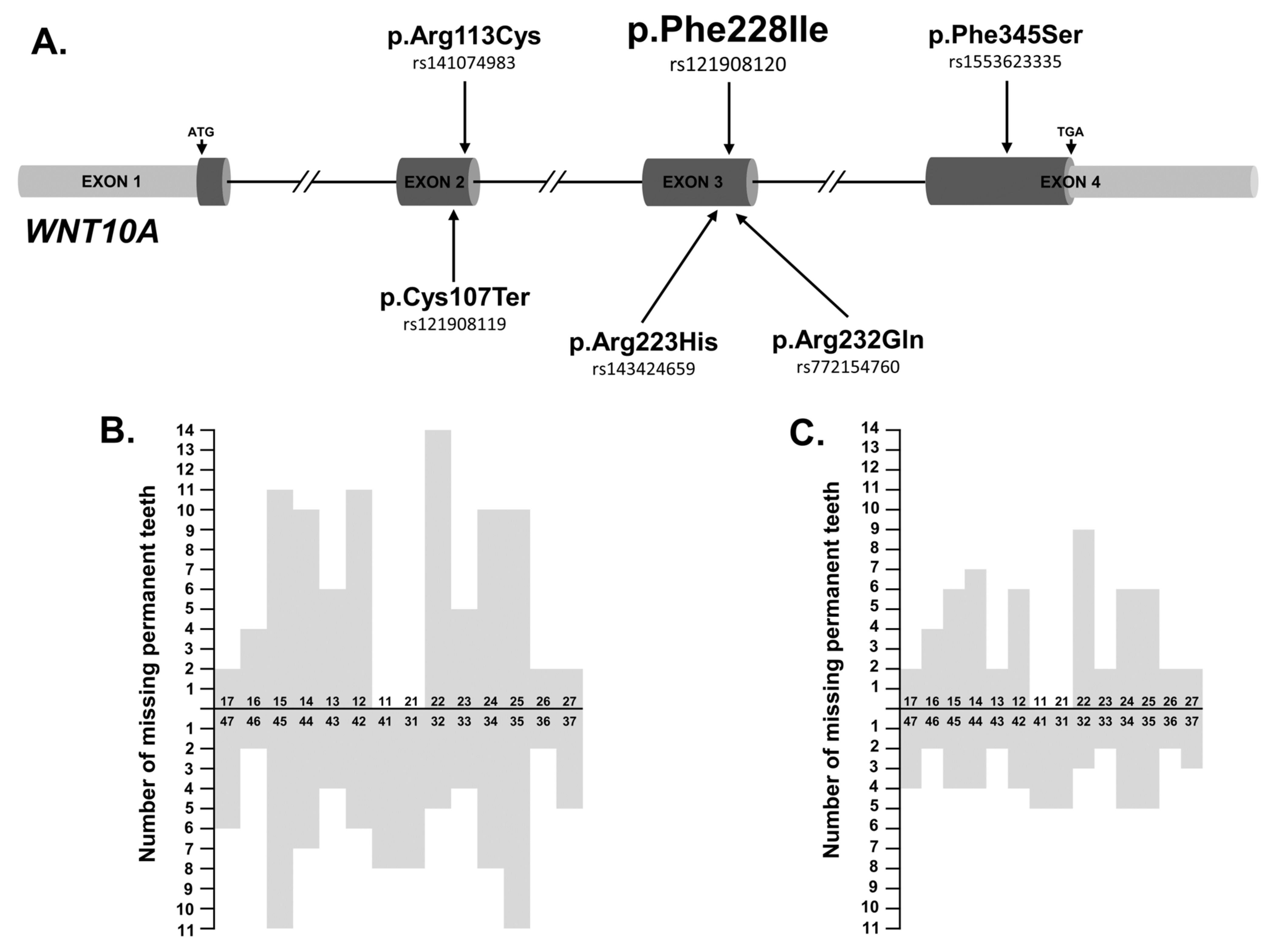
JCM, Free Full-Text
Filippi Syndrome disease: Malacards - Research Articles, Drugs, Genes, Clinical Trials

PDF) An unusual presentation of Rubinstein-Taybi Syndrome with bilateral postaxial polydactyly Corresponding author

Hennekam Lymphangiectasia-Lymphedema Syndrome 1 disease: Malacards - Research Articles, Drugs, Genes, Clinical Trials
DysmorphicNeonate: An Approach to Diagnosis in The Current Era
Identification of de novo EP300 and PLAU variants in a patient with Rubinstein–Taybi syndrome-related arterial vasculopathy and skeletal anomaly

1q21 1 Deletion Syndrome: Most Up-to-Date Encyclopedia, News & Reviews
Cureus, Whole-Exome Sequencing Identified a Novel DYRK1A Variant in a Patient With Intellectual Developmental Disorder, Autosomal Dominant 7
Genes, Free Full-Text

Medical Publications – Bohring-Opitz Syndrome

Skeletal Dysplasias - Endotext - NCBI Bookshelf
Recomendado para você
-
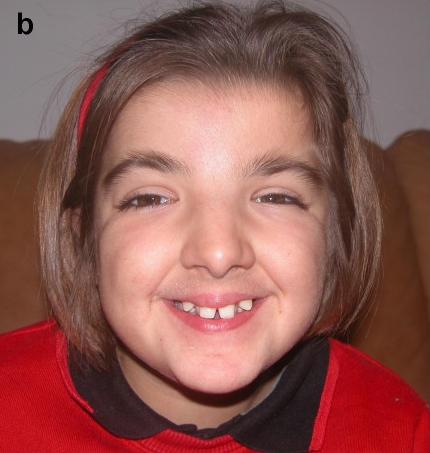 Forgotten Diseases Research Foundation11 novembro 2024
Forgotten Diseases Research Foundation11 novembro 2024 -
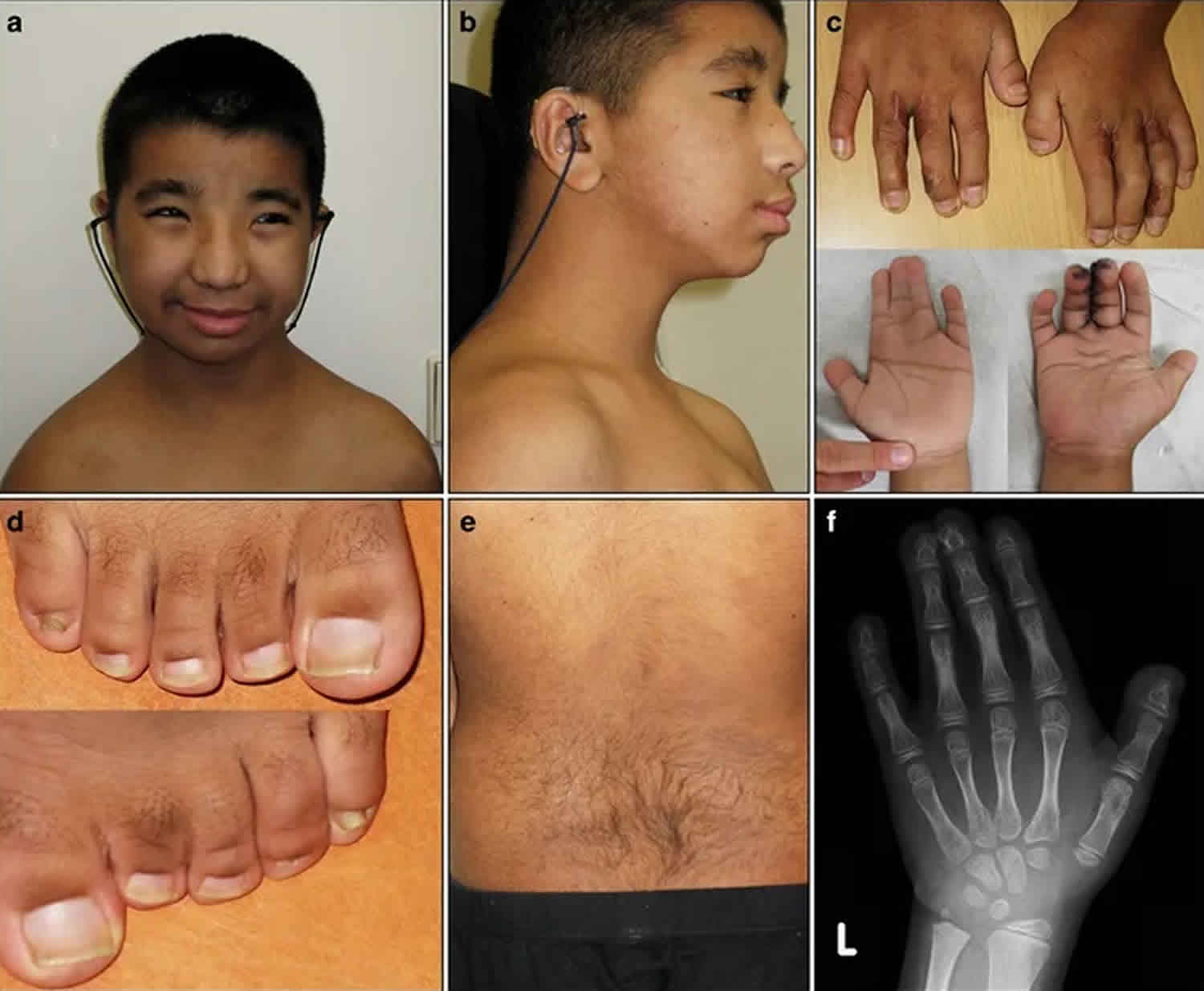 Rubinstein Taybi syndrome causes, symptoms, diagnosis, treatment & prognosis11 novembro 2024
Rubinstein Taybi syndrome causes, symptoms, diagnosis, treatment & prognosis11 novembro 2024 -
 A novel mutation c.4003 G>C in the CREBBP gene in an adult female with Rubinstein–Taybi syndrome presenting with subtle dysmorphic features - Li - 2010 - American Journal of Medical Genetics Part11 novembro 2024
A novel mutation c.4003 G>C in the CREBBP gene in an adult female with Rubinstein–Taybi syndrome presenting with subtle dysmorphic features - Li - 2010 - American Journal of Medical Genetics Part11 novembro 2024 -
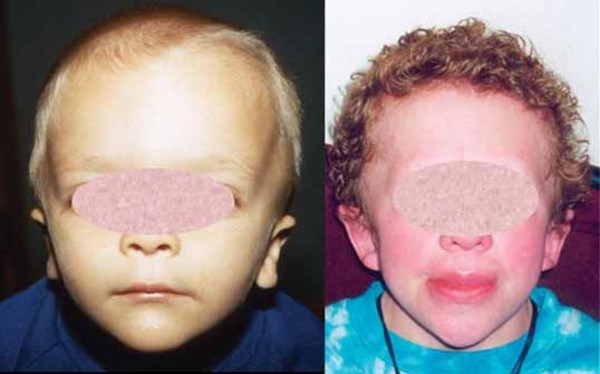 Floating-Harbor syndrome: MedlinePlus Genetics11 novembro 2024
Floating-Harbor syndrome: MedlinePlus Genetics11 novembro 2024 -
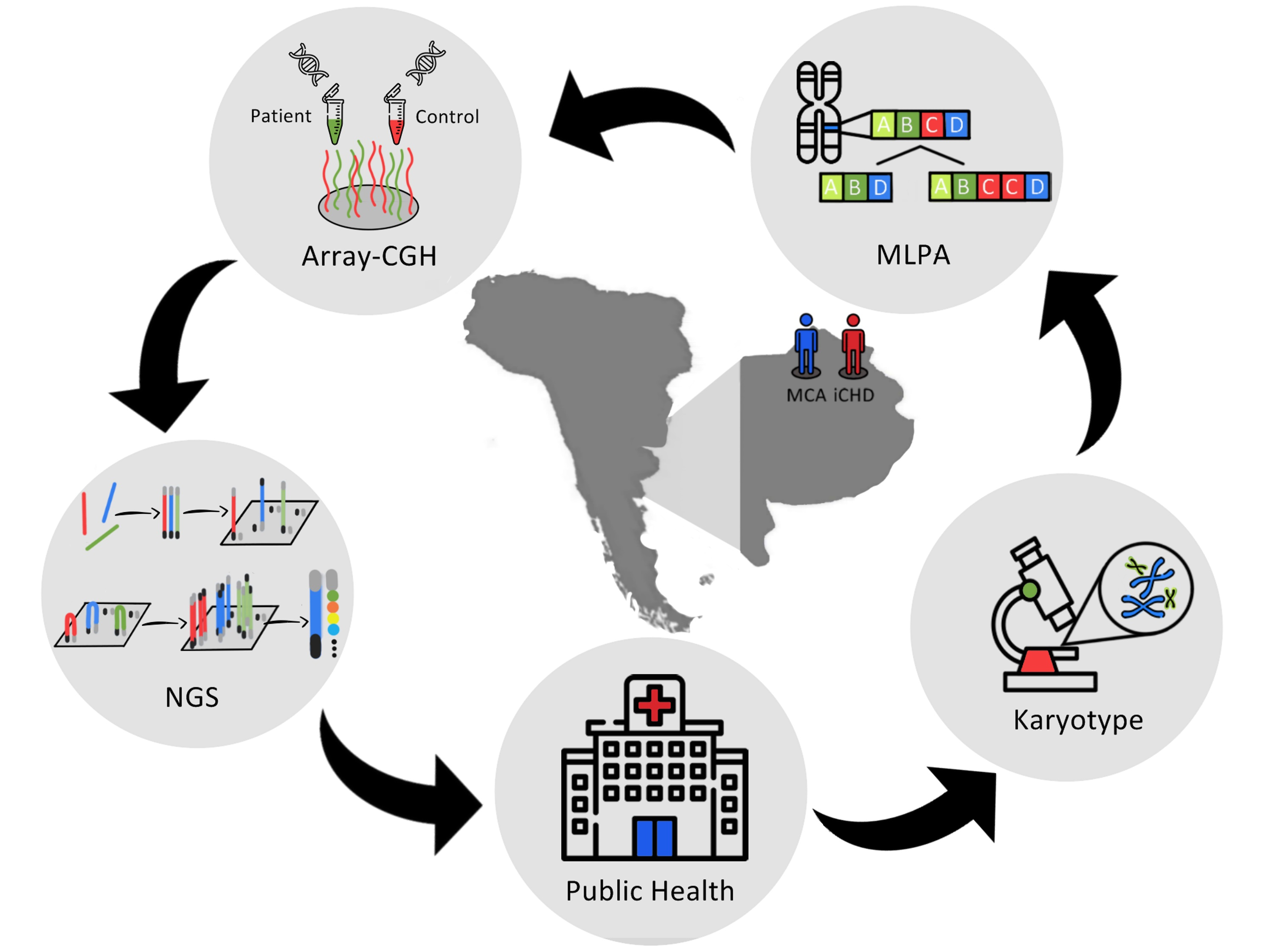
 PDF) Rubinstein-Taybi syndrome in diverse populations11 novembro 2024
PDF) Rubinstein-Taybi syndrome in diverse populations11 novembro 2024 -
JCM, Free Full-Text11 novembro 2024
-
 What is CdLS? Ben and his Brothers: Life with 4 boys and CdLS11 novembro 2024
What is CdLS? Ben and his Brothers: Life with 4 boys and CdLS11 novembro 2024 -
 Rubinstein–Taybi syndrome in diverse populations - Tekendo‐Ngongang - 2020 - American Journal of Medical Genetics Part A - Wiley Online Library11 novembro 2024
Rubinstein–Taybi syndrome in diverse populations - Tekendo‐Ngongang - 2020 - American Journal of Medical Genetics Part A - Wiley Online Library11 novembro 2024 -
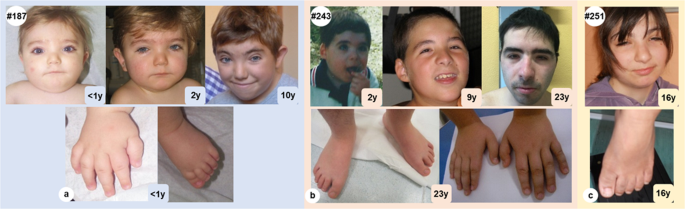 Expanding the phenotype associated to KMT2A variants: overlapping clinical signs between Wiedemann–Steiner and Rubinstein–Taybi syndromes11 novembro 2024
Expanding the phenotype associated to KMT2A variants: overlapping clinical signs between Wiedemann–Steiner and Rubinstein–Taybi syndromes11 novembro 2024 -
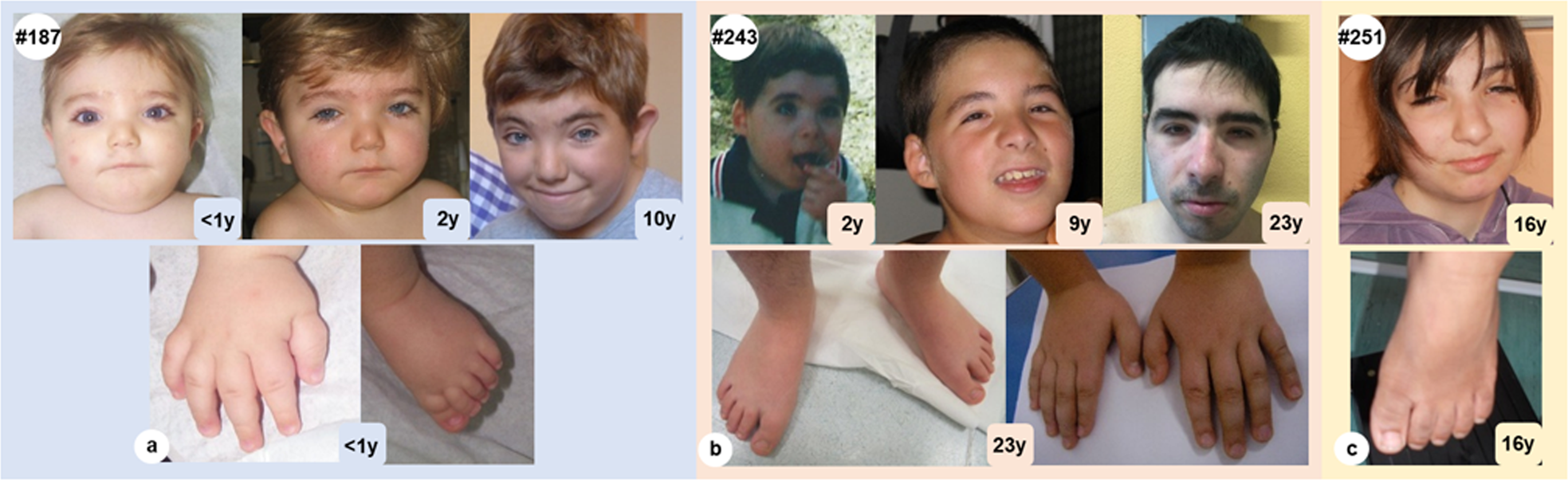 Expanding the phenotype associated to KMT2A variants: overlapping11 novembro 2024
Expanding the phenotype associated to KMT2A variants: overlapping11 novembro 2024
você pode gostar
-
 Play Subway Surfers Mumbai Free Online Games. KidzSearch.com11 novembro 2024
Play Subway Surfers Mumbai Free Online Games. KidzSearch.com11 novembro 2024 -
 ちっぽけな僕ら — World Trigger Ch160 'Hus 4′ Translation11 novembro 2024
ちっぽけな僕ら — World Trigger Ch160 'Hus 4′ Translation11 novembro 2024 -
 Juventus Fc Stock Illustrations – 43 Juventus Fc Stock Illustrations, Vectors & Clipart - Dreamstime11 novembro 2024
Juventus Fc Stock Illustrations – 43 Juventus Fc Stock Illustrations, Vectors & Clipart - Dreamstime11 novembro 2024 -
👨💻🔥 [New!] Hacker Tycoon 2🔥👨💻 - Roblox11 novembro 2024
-
 GTA 6: Confira as referências da vida real no trailer do jogo11 novembro 2024
GTA 6: Confira as referências da vida real no trailer do jogo11 novembro 2024 -
 Detailed Victorian House : r/Bloxburg11 novembro 2024
Detailed Victorian House : r/Bloxburg11 novembro 2024 -
 Boneca Miraculous Ladybug Baby Brink Com Ioiô 55 Cm - Novabrink - Bonecas - Magazine Luiza11 novembro 2024
Boneca Miraculous Ladybug Baby Brink Com Ioiô 55 Cm - Novabrink - Bonecas - Magazine Luiza11 novembro 2024 -
 Underfell Sans, Wiki11 novembro 2024
Underfell Sans, Wiki11 novembro 2024 -
Paradise Hotel11 novembro 2024
-
 Camiseta masculina Desenho Motor V8 Carro Diagrama Camisa Blusa Branca Estampada no Shoptime11 novembro 2024
Camiseta masculina Desenho Motor V8 Carro Diagrama Camisa Blusa Branca Estampada no Shoptime11 novembro 2024
![👨💻🔥 [New!] Hacker Tycoon 2🔥👨💻 - Roblox](https://tr.rbxcdn.com/10b1a5c6a33342e750a2efbdeedce234/500/280/Image/Jpeg)
